
Transkript
Seltene Krankheiten
Spinale Muskelatrophie
Genetische Abklärung bei SMA-Verdacht rasch veranlassen
Die spinale Muskelatrophie (SMA) ist die häufigste genetisch bedingte Todesursache im Säuglingsalter. Anders als früher ist die SMA mithilfe neuer Medikamente zu einer prinzipiell behandelbaren Erkrankung geworden. Im Folgenden werden die Grundlagen der neuen therapeutischen Ansätze zusammengefasst.
Die spinale Muskelatrophie (SMA) beruht auf einer autosomal rezessiv vererbten Mutation, die zu einem Verlust von Motoneuronen und progre dienter Muskelschwäche führt. Die Inzidenz der
Alter bei Manifestation der
Symptome
6 Monate
18 Monate
18 Jahre
SMA beträgt 1:6000 bis 1:10 000, die Prävalenz der Gen
träger in der Bevölkerung wird auf 1:40 bis 1:60 ge schätzt.
Typ 1
Typ 2
Typ 3
Typ 4
Die SMA ist die häufigste genetisch bedingte Todesursache
im Säuglingsalter. Säuglinge mit SMA weisen eine mus kuläre Hypotonie (floppy infant) mit voranschreitender
Anzahl SMN2-Kopien 1
2
Muskelschwäche auf. Die Mimik ist nicht beeinträchtigt.
3
Auch die Intelligenz ist normal. Symptome mit Verdacht auf SMA sind Zungenfaszikulationen, Tremor, verminder
4 ≥5
ter Hustenstoss, Schluckstörungen (bulbäre Muskel
schwäche) und paradoxe Atmung.
Abbildung: Phänotypen der spinalen Muskelatrophie (SMA)
Unterschiedliche Formen der SMA
(nach Schorling et al., 2020)
Das Spektrum der SMA reicht von schweren Verläufen,
die sich in den ersten Lebensmonaten manifestieren und denen sich die Erkrankung erst im Erwachsenenalter
rasch zum Tode führen, bis zu seltenen, verhältnismässig zeigt. Die Lebenserwartung ist bei Typ-3- und Typ-4-SMA
milden Auswirkungen des Gendefekts, die erst im Er offenbar nicht beeinträchtigt.
wachsenenalter sichtbar werden. Man definiert verschie Einige Autoren definieren darüber hinaus einen Typ 0
dene Typen der SMA, wobei die Übergänge fliessend sind (pränatale SMA); diese Kinder sterben in den ersten Le
(s. Abbildung).
benstagen.
Bei den meisten Patienten (50–60%) handelt es sich um
den Typ 1: Die SMA manifestiert sich in den ersten 6 Le Welche Mutation verursacht SMA?
bensmonaten. Diese Patienten werden nie frei sitzen Ursache der SMA ist in fast allen Fällen (95%) eine Dele
können. Die Lebenserwartung beträgt für 90 Prozent der tion und nur selten eine Punktmutation im SMN1-Gen
Betroffenen ohne Behandlung weniger als 2 Jahre.
auf Chromosom 5q. SMN1 steht für «survival of moto
Bei etwa einem Drittel der SMA-Patienten treten die ers neuron 1». Das vom SMN1 kodierte SMN-Protein ist es
ten Symptome im Alter von 6 bis 18 Monaten auf. Sie senziell für das Überleben der Motoneuronen. Der Defekt
erlernen das freie Sitzen, werden aber nie ohne Hilfe in SMN1-Gen führt zu einem Mangel an SMN-Protein
gehen können (Typ 2). Die Überlebensrate bei Typ-2-SMA und letztlich zur SMA.
beträgt 10 Jahre nach Erkrankungsbeginn für mehr als Man weiss nicht genau, wie das SMN-Protein das Über
90 Prozent der Patienten.
leben der Motoneuronen bewirkt. Das SMN-Protein
Rund 10 Prozent der SMA-Patienten gehören dem Typ 3 kommt im gesamten Organismus vor und ist Teil des zel
an. Sie entwickeln erste Symptome im Alter über 18 Mo lulären Spliceosoms, desjenigen RNA-Protein-Komplexes,
naten beziehungsweise erst im Kindes- und Jugendalter der aus der ersten «Abschrift» der genetischen Informa
und können zumindest zeitweise ohne Hilfe gehen. Sehr tion die für die Proteinsynthese überflüssigen und stören
gering ist der Anteil an SMA-Patienten von Typ 4, bei den Abschnitte entfernt (splicing).
3/20 Pädiatrie
29
Seltene Krankheiten
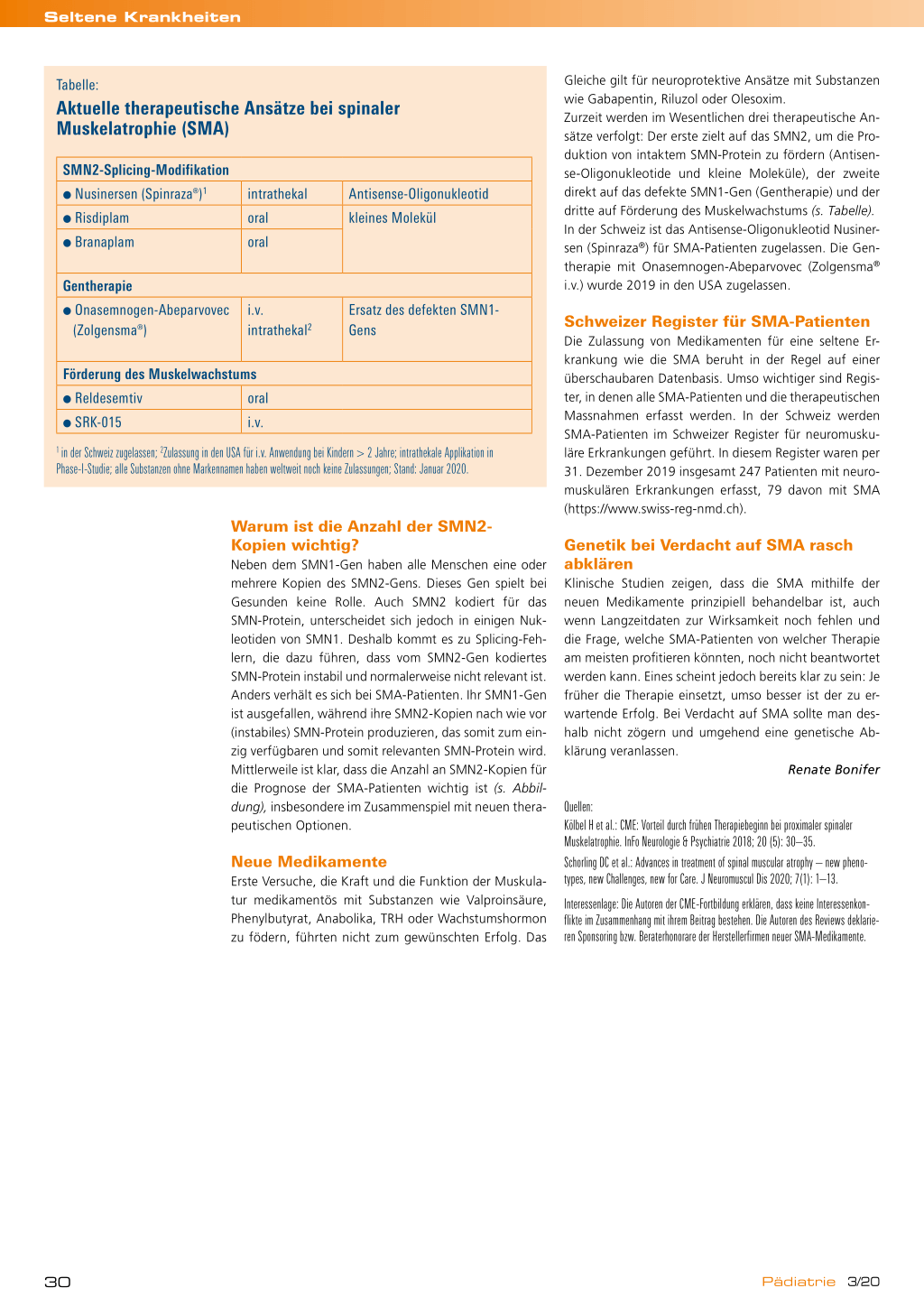
Tabelle:
Aktuelle therapeutische Ansätze bei spinaler Muskelatrophie (SMA)
SMN2-Splicing-Modifikation ● Nusinersen (Spinraza®)1 ● Risdiplam ● Branaplam
intrathekal oral oral
Antisense-Oligonukleotid kleines Molekül
Gentherapie
● Onasemnogen-Abeparvovec (Zolgensma®)
i.v. intrathekal2
Ersatz des defekten SMN1Gens
Förderung des Muskelwachstums
● Reldesemtiv
oral
● SRK-015
i.v.
1 in der Schweiz zugelassen; 2Zulassung in den USA für i.v. Anwendung bei Kindern > 2 Jahre; intrathekale Applikation in Phase-I-Studie; alle Substanzen ohne Markennamen haben weltweit noch keine Zulassungen; Stand: Januar 2020.
Warum ist die Anzahl der SMN2Kopien wichtig?
Neben dem SMN1-Gen haben alle Menschen eine oder mehrere Kopien des SMN2-Gens. Dieses Gen spielt bei Gesunden keine Rolle. Auch SMN2 kodiert für das SMN-Protein, unterscheidet sich jedoch in einigen Nuk leotiden von SMN1. Deshalb kommt es zu Splicing-Feh lern, die dazu führen, dass vom SMN2-Gen kodiertes SMN-Protein instabil und normalerweise nicht relevant ist. Anders verhält es sich bei SMA-Patienten. Ihr SMN1-Gen ist ausgefallen, während ihre SMN2-Kopien nach wie vor (instabiles) SMN-Protein produzieren, das somit zum ein zig verfügbaren und somit relevanten SMN-Protein wird. Mittlerweile ist klar, dass die Anzahl an SMN2-Kopien für die Prognose der SMA-Patienten wichtig ist (s. Abbildung), insbesondere im Zusammenspiel mit neuen thera peutischen Optionen.
Neue Medikamente
Erste Versuche, die Kraft und die Funktion der Muskula tur medikamentös mit Substanzen wie Valproinsäure, Phenylbutyrat, Anabolika, TRH oder Wachstumshormon zu födern, führten nicht zum gewünschten Erfolg. Das
Gleiche gilt für neuroprotektive Ansätze mit Substanzen wie Gabapentin, Riluzol oder Olesoxim. Zurzeit werden im Wesentlichen drei therapeutische An sätze verfolgt: Der erste zielt auf das SMN2, um die Pro duktion von intaktem SMN-Protein zu fördern (Antisen se-Oligonukleotide und kleine Moleküle), der zweite direkt auf das defekte SMN1-Gen (Gentherapie) und der dritte auf Förderung des Muskelwachstums (s. Tabelle). In der Schweiz ist das Antisense-Oligonukleotid Nusiner sen (Spinraza®) für SMA-Patienten zugelassen. Die Gen therapie mit Onasemnogen-Abeparvovec (Zolgensma® i.v.) wurde 2019 in den USA zugelassen.
Schweizer Register für SMA-Patienten
Die Zulassung von Medikamenten für eine seltene Er krankung wie die SMA beruht in der Regel auf einer überschaubaren Datenbasis. Umso wichtiger sind Regis ter, in denen alle SMA-Patienten und die therapeutischen Massnahmen erfasst werden. In der Schweiz werden SMA-Patienten im Schweizer Register für neuromusku läre Erkrankungen geführt. In diesem Register waren per 31. Dezember 2019 insgesamt 247 Patienten mit neuro muskulären Erkrankungen erfasst, 79 davon mit SMA (https://www.swiss-reg-nmd.ch).
Genetik bei Verdacht auf SMA rasch abklären
Klinische Studien zeigen, dass die SMA mithilfe der neuen Medikamente prinzipiell behandelbar ist, auch wenn Langzeitdaten zur Wirksamkeit noch fehlen und die Frage, welche SMA-Patienten von welcher Therapie am meisten profitieren könnten, noch nicht beantwortet werden kann. Eines scheint jedoch bereits klar zu sein: Je früher die Therapie einsetzt, umso besser ist der zu er wartende Erfolg. Bei Verdacht auf SMA sollte man des halb nicht zögern und umgehend eine genetische Ab klärung veranlassen.
Renate Bonifer
Quellen: Kölbel H et al.: CME: Vorteil durch frühen Therapiebeginn bei proximaler spinaler Muskelatrophie. InFo Neurologie & Psychiatrie 2018; 20 (5): 30–35. Schorling DC et al.: Advances in treatment of spinal muscular atrophy – new pheno types, new Challenges, new for Care. J Neuromuscul Dis 2020; 7(1): 1–13.
Interessenlage: Die Autoren der CME-Fortbildung erklären, dass keine Interessenkon flikte im Zusammenhang mit ihrem Beitrag bestehen. Die Autoren des Reviews deklarie ren Sponsoring bzw. Beraterhonorare der Herstellerfirmen neuer SMA-Medikamente.
30
Pädiatrie 3/20